seismic wf
Purpose
seismic wf runs the entire SEISMIC-RNA pipeline end-to-end, from FASTQ
files (or any later-stage input) through alignment, mutation identification,
filtering, clustering, table generation, structure prediction, graphing, and
optional export.
It is the only command most users need to invoke.
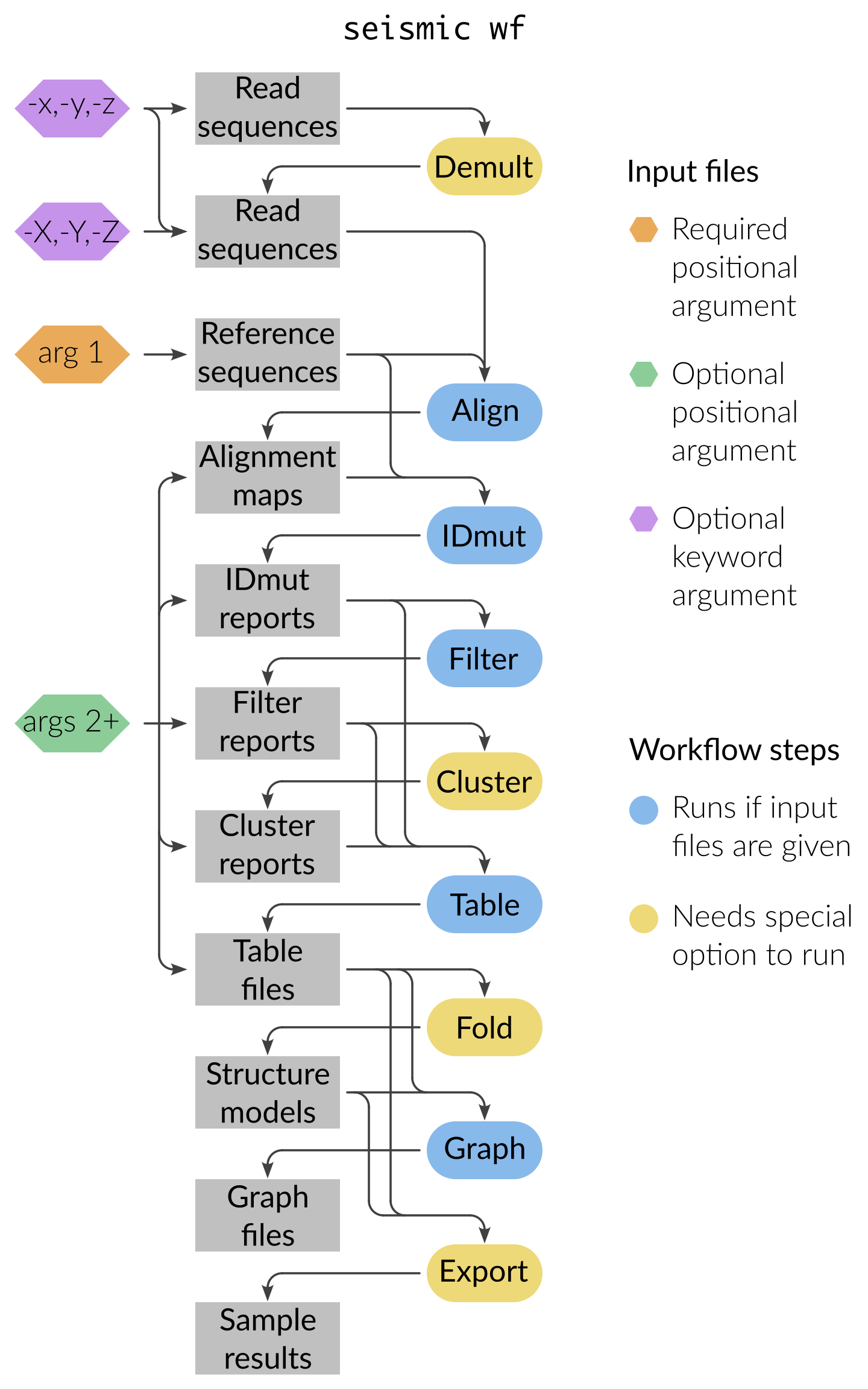
For each input file, wf resumes the pipeline from the step that produces
that file and runs everything downstream.
For example, a FASTQ goes through every step; a Filter report goes only
through Fold/Export/Graph.
Note
By default, the Cluster, Fold, Draw, and Export steps do not run.
Turn them on with --cluster, --fold, --draw, and --export
respectively.
Inputs
- Required
A FASTA file of one or more reference sequences (first positional argument; see FASTA: Reference sequences).
- Reads (any combination, optional)
-xpaired-end reads in two separate FASTQ files-ypaired-end reads interleaved in one FASTQ file-zsingle-end reads-X/-Y/-Zsame as above but for already-demultiplexed reads
- Intermediate files (any combination, optional)
Given as positional arguments after the FASTA:
SAM / BAM / CRAM alignment maps (see SAM, BAM, and CRAM: Alignment Maps)
IDmut, Filter, Cluster, or Table reports (see Report Formats)
Table CSVs (skip directly to Fold / Export / Graph)
See Specify Input Files for ways to list many input files at once (glob patterns, directory recursion, mixed positional and keyword inputs).
Outputs
seismic wf runs each step into the output directory given by
--out-dir (default ./out).
Most output paths are structured {out}/{sample}/{step}/{ref}/{region}/....
Refer to each step for the specific output structure: seismic align,
seismic idmut, seismic filter, seismic cluster, seismic table, seismic fold,
seismic graph, seismic draw, seismic collate, seismic export.
Quick example
Run the entire pipeline on a paired-end FASTQ pair against refs.fa:
seismic wf -x sampleA_R1.fq -x sampleA_R2.fq refs.fa
Run with clustering, folding, and export turned on:
seismic wf --cluster --fold --export -x sampleA_R1.fq -x sampleA_R2.fq refs.fa
Resume from existing alignment maps and existing IDmut reports:
seismic wf refs.fa out/sampleA/align/refX.bam out/*/idmut/refY/idmut-report.json
Options
- Optional steps
--cluster/--no-cluster— run the Cluster step (default: off).--fold/--no-fold— run the Fold step (default: off).--draw/--no-draw— run the Draw step (default: off).--export/--no-export— run the Export step (default: off).--max-clusters/-k— maximum number of clusters to try once the Cluster step runs (default: 0, i.e. no limit). Giving-ka positive number also turns on the Cluster step.
- Per-step options
Every option accepted by any individual pipeline command can be given to
seismic wf, except for--branch(use--wf-branchinstead; see Branches below). For the complete list of options and what each does, see the per-step pages (seismic align, seismic idmut, seismic filter, seismic cluster, seismic table, seismic fold, seismic graph, seismic draw, seismic collate, seismic export) and the auto-generated Command Line Reference.- Branches
--wf-branch STEP NAMERun one step of the workflow under a branch, writing its outputs to
{out}/{sample}/{STEP}_{NAME}/instead of the default directory. Give the step name followed by the branch name, and repeat the option to branch several steps, e.g.--wf-branch filter strict --wf-branch cluster strict.STEPmust be one ofdemult,align,idmut,filter,filterscan,cluster,clusterscan, orfold; any other name raises an error. See Branches.
- Global options
--out-dir— output directory (default./out).--num-cpus— number of parallel processes (see Parallelize Tasks).--force— overwrite existing output files.
Options and positional arguments can be mixed freely on the command line.
Caveats
The optional steps (Cluster, Fold, Draw, Export) are off by defaults.
The FASTA file must contain the same references as those used for any pre-existing BAM files or reports given as input. Mismatched references cause the IDmut step to fail; see seismic idmut.
All inputs and outputs share a single
--out-dir. Re-runningwfagainst the same--out-dirwithout--forcewill skip steps whose outputs already exist (issuing a warning for each existing output file).
Performance tips
Use
--num-cpus Nto parallelize across reads and references; see Parallelize Tasks.For very large datasets, tune
--batch-size(see seismic idmut).Resume a partially-completed run by re-invoking
wfwith the same--out-dir; only missing outputs are produced.
Common errors
seismic wf delegates to the individual pipeline commands, so its error
messages come from whichever step failed.
See the per-step pages for the exceptions each step can raise:
seismic align — alignment errors (missing reference, Bowtie2 failure)
seismic idmut — reference mismatch, insufficient reads, mapping quality
seismic filter — region/probe errors
seismic cluster — convergence and clustering failures
seismic fold — RNAstructure
DATAPATHnot set (see Install)
Common unexpected results
- No clustered outputs
You forgot
--cluster.- No structure models
You forgot
--fold.- No webapp JSON
You forgot
--export.- Empty output for some references
The Align step deletes BAM files that contain fewer than
--min-readsreads (default 1000) to prevent cluttering the output directory with BAM files that are too small to be useful in downstream steps.wfdid not run a step you expectedConfirm what inputs you passed.
wfonly runs the steps that recognize an input file you provided. For example, passing a table CSV file from the IDmut step (idmut-pos-table.csv) will not trigger the Filter step because Filter only accepts IDmut report JSON files (idmut-report.json).
See also
Specify Input Files — how to list many input files
Parallelize Tasks — how parallelism works
Log Messages — controlling verbosity and log files
Branches — run variant analyses under a different name
How SEISMIC-RNA Works — visual tour of each pipeline step
Tutorials — step-by-step worked examples